

地址:廣州市南沙區大崗鎮智新一路8號聯東U谷廣州南沙國際企業港7棟5樓
產品咨詢:13392693259
耗材電話:18998328368
售后服務:020-87684117
傳 真:020-87684846
Email:info@gdana.com
網址:http://www.beehive-home.cn/
2021.06.16
硒(Seleniun,Se)是生命活動中必需的微量元素,在人體中以硒蛋白的形式存在,具有提高免疫、抗氧化、抗腫瘤、延緩衰老、解毒和抗輻射等生物與生理功能。人體硒缺乏會降低硒蛋白的表達及生物學功能的正常發揮,會引起如貧血、冠心病、高血壓、癌癥等40多種疾病,適量補硒有利于人體健康[1-3]。但硒對人體的有益生理功能必需控制在安全濃度范圍之內,補硒過量又會引起生物體中毒[4],世界衛生組織(WHO)、聯合國糧農組織(FAO)、國際原子能機構(IAEA)均規定了明確指標:膳食硒的供給量為50~250μg?d-t,成人個體硒安全攝入量為400g?d-1[5]。人體從環境中攝取硒的主要途徑是食物,食物鏈中硒又主要來源于植物,因此對植物硒的準確測定顯得十分重要。

1、實驗部分
1.1方法
目前測定硒的方法主要有光學分析法(分光光度法、熒光法、原子熒光光譜法,原子吸收法等)6[6-8]、電化學分析法(陰極溶出伏安法、極譜法等)[9,10]、色譜學分析法(離子色譜法、高效液相色譜法、氣相色譜法等)[11]、聯用技術(電感耦合等離子體質譜法)等[12,13],每種方法都有其優缺點,在不同的情況下可選擇適當的檢測方法進行分析。其中氫化物發生-原子熒光光譜法(HG-AFS)因其測定硒具有較高的靈敏度和精確度,所用試劑毒性小,操作簡便,實用性強,已入選食品安全國家標準“食品中硒的測定(GB5009.93-2010)”的第一法[14],尤其適合痕量硒的測定,本工作采用氫化物發生-原子熒光光譜法測定植物樣品中的硒。
植物硒多以有機形態存在,儀器測定前需轉化為無機離子,目前處理植物樣品的方法有干灰法和濕法消解等方法,鑒于硒的易揮發特性,植物硒多采用濕法消解。“國標法-食品中硒的測定(GB 5009.93-2010)”采用兩種濕消解法:硝酸十高氯酸“電熱板”敞開體系常壓消解法,硝酸十雙氧水“微波”密閉體系消解法。“電熱板”消解為傳統濕消解法,簡便成本低,易操作,但試劑消耗多,溫度不能精確控制,消解時多憑經驗,消解溫度過高或時間過長極易造成硒損失,溫度過低有機硒又不能完全被消解,導致測定結果偏低。微波消解是目前被廣為推薦的一種微量元素消解方法,程序控溫,氧化劑用量少,微波消解時間短且完全,但缺點是微波消解后還需在電熱板上趕走殘留余酸才能確保測量的準確性。硒的易揮發性決定了硒必須低溫趕酸,這一步驟所需時間較長,總體消化過程中只是節省了微波消解部分的時間,且單個樣品消化量較少,通常只有0.1~0.5g。本法采用程序控溫的石墨消解儀消解處理植物硒,單個樣品消化量可達10.0g以上,可以滿足痕量樣品的大稱樣量的分析要求。石墨消解雖屬于敞開體系,但具有微波消解儀相同的自動控溫程序,裝有試樣的消解管全部包裹于石墨加熱孔中,能量不易散失,加熱效率高且均勻,消解趕酸能同時進行。且石墨消解管還自帶刻度,消解完成后無需轉移樣液可直接定容,有別于電熱板和微波消解的二次轉移重新定容過程,避免了在轉移過程中的污染和損失。特別是儀器價格比微波消解儀低,經濟適用,常規實驗室都有能力購買,操作簡便安全,所用試劑毒性小,一次可處理大批樣品,尤其適合植物樣品批量分析,其測定結果令人滿意。
1.2儀器與試劑
格丹納DS-360智能石墨消解儀;雙道原子熒光光譜儀,高性能硒空心陰極燈(北京有色金屬研究總院);超純水機,氬氣:純度為99,99%。
硝酸(HNO3)、高氯酸(HClO)、鹽酸(HCl)均為GR。硼氫化鉀溶液(10.0g?L-1):稱取5.0g氫氧化鉀(KOHAR)溶于約500mL純水中,加入10.0g硼氫化鉀(KBH4CP),緩緩搖動使其溶解,定容至1000mL,混勻即可,現配現用。鐵氰化鉀(100g?L-1)溶液:稱取10.0g鐵氰化鉀(K3Fe(CN)6),AR,溶于100mL純水中,混勻。植物標樣為灌木葉(GBW07602/GSV-1)、柑橘葉(GBW10020/GSB-11)、茶葉(GBW10016/GSB7)、圓白菜(GBW10014/GSB-5),大米(GBW10010/GSB-1)分別購自地球物理地球化學勘査硏究所。
硒標準儲備液溶液(100mg?L-1),硒質控環境標準溶液(GSBZ50031-94(203710)),購自中國環境保護部標準樣品研究所。試驗中純水為18.3MΩ?cm(25C)超純水。
1.3樣品與測試液的制備
稱取2.0g(精確至0.0001g)洗凈60C烘干粉碎的植物樣,加入10.0mL硝酸十高氯酸(8+2)(g)混酸及幾粒玻璃珠于100mL刻度消解管中,上端放彎頸玻璃小漏斗便于回流,混勻放置在石墨消解儀的石墨加熱孔中,于通風良好的通風櫥內冷消化過夜12h后,石墨消解儀會按預先設定的消解程序進行升溫消化,升溫程序為:室溫→50C保持0.5h100C保持1h-→160C保持2h→-180C保持,保持時間視樣品消化情況而定。其間要觀察消化液顏色,若較深,及時補加硝酸,反復多次,直到沒有棕色氮氧化物氣體冒出。當溶液變為清亮無色并伴有濃白煙岀現時,繼續保持180C加熱,其間可加入1~2mL純水1~2次,目的是趕殘余酸,這樣可以解決濕法消解測定空白高的特點,直至加熱消化到剩余體積為2mL左右時,切不可蒸干,取下冷卻至室溫,再加入5.0mL50%HCl(g)于剩余消化液中,再次至于消解儀中升溫至100C(或沸水浴加熱),保持10~15mn,溶液變為清亮無色并伴有白煙時,逐將Se+還原成Se+。取出冷卻用純水定容至25mL,密封搖勻備用。同時做試劑空白。
測試液的制備:按所需稀釋比例吸取以上待測液的上清液1.0~5,0mL至10mL比色管中,加入適量的50%HCl(g)(以確保定容后的樣液酸度為15%),1.0mL鐵氰化鉀(10gg?L-1)溶液,純水定容并混勻。靜置30min后上機測試。
1.4硒標準溶液配制
將100mg?L-1硒標準儲備液用5%HCl溶液稀釋至100gg?L標準應用液。從100gg?L硒標準應用液中,分別移取0.00,0.50,1.00,2.00,3.00,4.00,5.00mL至7個50mL容量瓶中,加入少量純水,再分別向每個容量瓶中加入50%HCl15.0mL(以確保定容后的樣液酸度為15%),5.0mL鐵氰化鉀(100g?L-1)溶液,純水定容,即可得到一組濃度為0.00,1.00,2.00,4.00,6.00,8.00,10.00gg?L-1系列硒標準工作溶液。
2、結果與討論
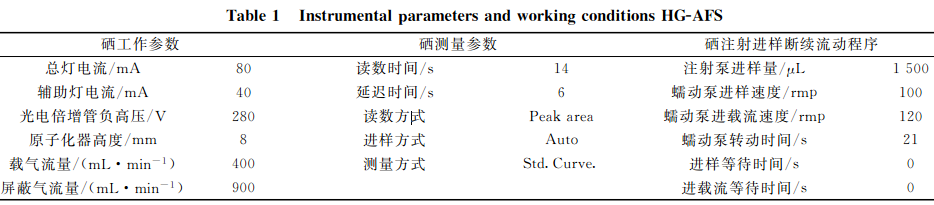
2.1原子熒光光譜儀工作參數
考慮燈電流、光電倍增管負高壓、載氣流量和反應介質等對分析靈敏度、精確度和準確度的影響,結合AFS-9700型雙道原子熒光光譜儀的儀器條件,經反復試驗,確定了植物硒測定的工作參數,特別對讀數時間和延遲時間進行了反復摸索,確定了讀數時間為14s,延遲時間必須為6s時,在儀器的信號采集單元內才能獲得一個完整正態峰型,此時峰面積大,達到了積分效果,說明在上述儀器工作條件下,硒熒光強度響應值高,結果見表1。
2.2消解液用量
準確稱取2.0mg(精確至0.0001g)植物標樣柑橘葉(GBW10020(GSB-11))樣品21份,分3組,每組7次重復。分別加入混酸比為HNO3+HCl0O4(V+V)=(6+4),(8+2)和(9+1)消解液10.0mL及幾粒玻璃珠冷消解12h過夜,消解處理制樣步驟同1.3,測試硒的相對熒光值,結果見表2。
可知:混酸比為HNO3+HClO(V+V)=8+2處理的柑橘葉樣品中硒的相對熒光值高,且試樣重復良好,其余兩組熒光值偏低,重復不穩定,特別是混酸比為HNO3+HCIO4(V+V)=6+4一組重復差異較大。說明用HNO3+HCO4(V+V)=8+2混酸比消解處理柑橘葉樣品時,硒的提取完全。
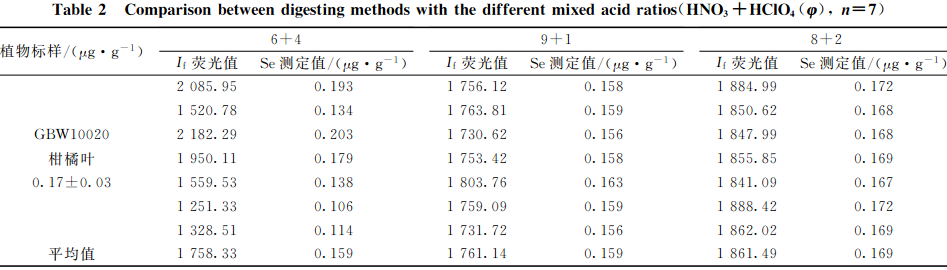
全植物樣品中含有大量有機質,在酸量較少的情況下HClO1遇到大量的有機物不僅易發生爆炸,還會造成空白值偏高。實驗中加入的HClO41分別為4.0,2.0,1.0mL。發現HClO加入量越多,消化液中HCIO4的殘留量就越多。如果消化至剩余體積的2mL左右時,HCO冒煙時間過長,硒有損失,則測定結果偏差較大;HClO44加入量少,消解速度慢,時間過短,消解液顏色深,有機質消解又不完全。對于稱樣量為2.0g的柑橘葉樣品來說,選擇混酸比HNO3HClO(V+V)=(8+2)mL,,HClO1加入量為2.0mL,就可以使有機質充分消解。當稱樣量大時,可以相應增加HClO的量,消解液中HClO)殘留量約為0.5~1.0mL較為合適。其他實驗條件的影響研究均采用(8十2)混酸比來消解植物樣品。
2.3消解溫度和消解時間的選擇
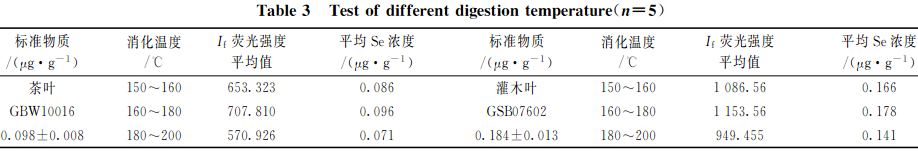
準確稱取2.0g(精確至0.0001g)植物標樣茶葉(GBW10016)樣品和灌木葉(GSB07602)樣品各15份,分3組,每組5次重復稱樣,以2.2中HNO3+HClO4(V+V)(8+2)mL混酸比溶液進行消化處理,再以不同的消解溫度進行消化處理,測試硒的相對熒光值,結果見表3。
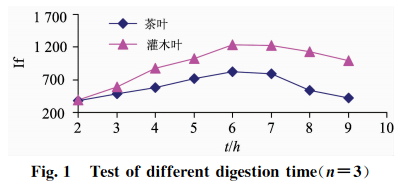
準確稱取2.0g(精確至0.0001g)植物標樣茶葉(GBW10016)樣品和灌木葉(GSB07602)樣品各24份,分8組,每組3次重復稱樣,以2.2中HNO3+HClO4(V+V)(8+2)mL混酸比溶液,以表3中的160~180C為消化溫度,分別設定8組消化時間為2,3,4,5,6,7,8,9h,測試硒的相對熒光值,結果見圖1。
由表3和圖1可知:消化溫度控制在160~180C之間消化時間在6~7h之間時,兩種植物樣品硒的提取完全;當消化溫度<160C,或>180C時,消化時間過短或過長時,硒消化不徹底或容易揮發,都會導致熒光值偏低且不穩定。
2.4還原劑硼氫化鉀(KBH4)和載流鹽酸(HCI)的影響
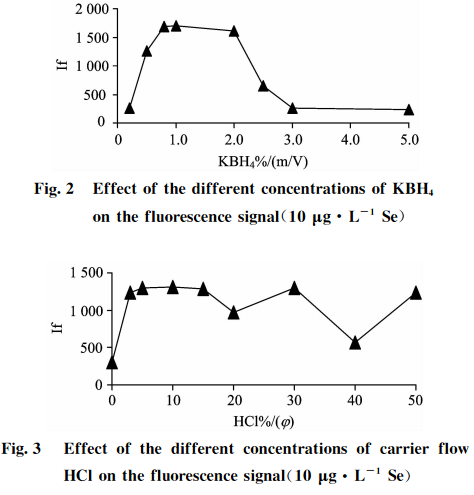
在儀器條件不變的情況下,測試10Hg?L-的Se標準溶液隨載流HCl和還原劑KBH4濃度變化對硒熒光強度的影響,結果見圖2和圖3。可知,當載流HCl和還原劑KBH4濃度低時熒光強度低,信號靈敏度差,說明還原能力弱;當載流HCl和還原劑KBH41濃度增大時,熒光強度也隨之增強;當兩者濃度太高時,反應產生的氫氣多,泡沫都易沖入管道,熒光強度反而會降低,這是由于過量的氫氣稀釋了火焰中硒原子蒸氣造成的。實驗結果表明:對于10pg?L1的Se標準溶液,還原劑KBH4濃度在0.8%~2.0%(m/V)之間,載流HC1濃度在4%~15%(g)之間,火焰平穩,熒光值達到穩定,信噪比和靈敏度高。綜合考慮選擇還原劑濃度為1.0%KBH4(m/V)+0.5%KOH(m/V),載流HC1為10%(g)。
所配還原劑KBH4溶液要含有一定濃度的KOH以保證溶液的穩定性。建議KOH的濃度為0.2%~0.5%(m/V),濃度過低,KBH4會分解,濃度過高則會影響氧化還原反應的總體酸度,離開KOH和KBH4的濃度來討論酸度是毫無意義的。
2.5樣品酸度(HC的選擇樣
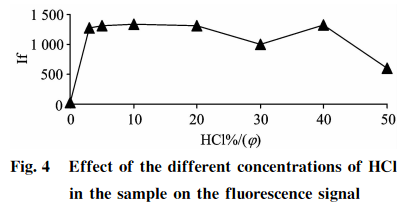
品消化過程的酸度確保了Se+還原成Se+,上機測試前樣品經稀釋后制備液酸度也會影響硒的測定結果。分別吸取100gg?L-標準應用液5.0mL于7個50mL的具塞比色管中,分別加入0.0,1.5,2.5,5.0,10.0,15.0,20.025,0mL濃HCl,用純水稀釋定容至刻度,用以制備不同酸度的10gg?L硒標準溶液,以2.4中的載流和還原劑濃度進行上機測定,結果見圖4。可知,扣除空白后,HCl(g)在3%~20%范圍內,10gg?L-1硒標準溶液具有較強且穩定的熒光強度;當樣品HCl(q)>20%時,硒熒光值受到抑制且不穩定,考慮到樣品酸度的提高有利于硒的還原,同時可以減少共存元素的干擾,故本方法選用15%HCl(g)酸度作為樣品和標準溶液上機測試時制備液的酸度,才能保證測量的穩定性和一致性。
2.6線性方程、檢出限
植物樣品硒的測定中,標準曲線范圍無需很大,本實驗在0~10!g?L-的濃度范圍內標準曲線線性相關很好,r=0.9999,回歸方程y=131.67x+203.94;又對0~100gL-濃度范圍進行了測定,其線性相關也很好,r=0.9997,回歸方程為y=132.43x+315.13,說明本方法不僅靈敏度高,而且線性范圍寬。
在本實驗條件下,連續測定空白熒光值11次,并以3倍的標準偏差除以工作曲線的斜率計算出溶液中硒的檢岀限0.018g?L-1,若以2.0g植物樣品定容25mL計算,樣品檢出限0.225gg?kg-1
2.7標準物質分析及回收率和精密度
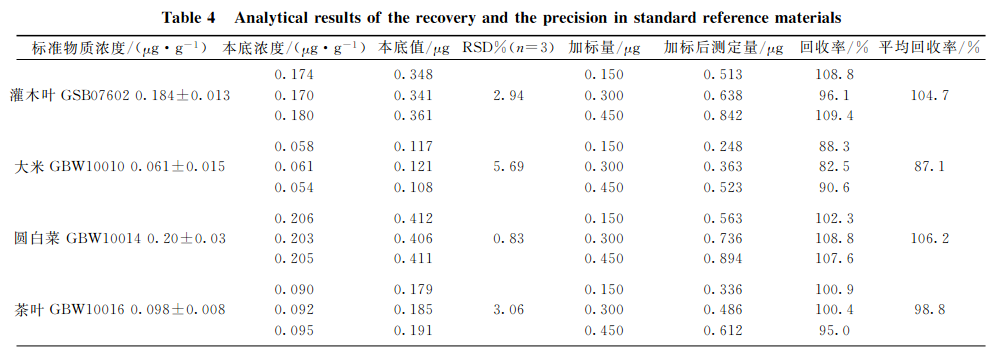
分別選取灌木葉、大米、圓白菜、茶葉和柑橘葉五種植物標準樣品,每種重復3次準確稱取2.0g(精確至0.0001g),按上述實驗條件進行消解,測定本底值。再對每種植物標樣稱取子樣,每種重復3次準確稱取2.0g(精確至0.0001g),此次每種試樣中分別加入1.0mg?L-1硒標準溶液150,300,450gL,進行消解并測定加標后值,同時測定硒質控環境標準溶液(GSBZ50031-94(203710)),結果見表4。可見,硒標準加標回收率在87.1%~106.2%之間,精密度(n=3)在0.83%~5.69%之間,所測結果均與標準物質的參考值吻合,完全達到分析要求。

3、結論
采用程序控溫石墨爐消解-氫化物熒光光諧法在本文得到的實驗條件下測定以柑橘葉、茶葉、灌木葉、圓白菜、大米等為代表的幾種植物樣品中的硒,線性范圍寬、靈敏度高、檢出限低、穩定性好的顯著特點,尤其適合這類批量植物樣品中硒的痕量分析,且該方法操作簡便安全,實用性強,儀器成本低,所用試劑毒性小,可作為一般實驗室的常規分析方法。
參考文獻
[1]FergusonLR,KarunasingheN,ZhuS,etal.MutationResearch/FundamentalandMolecularMechanismsofMutagenesis,2012,733(1-2)
[2]WANGKui(王夔).Traceelementsinlifescience,2ndedition(生命科學中的微量元素,第2版)Beijing:ChinesemetrologyPress(北京:中國計量出版社),1996.650
[3]BAOJian-min,YUXiao-yan,LIUWei,etal(包建民,于曉燕,劉微,等).JournalofInstrumentalAnalysis(分析測試學報),2012,31(7):804
[4]ReideME,StrattonbMS,LillicocAJ,etal.JournalofTraceElementsinMedicineandBiology,2004,18(1):68
[5]MAXiu-jie,ZHANGYUE-an(馬秀杰,張躍安).ChinesejournalofPublicHealth(中國公共衛生),2009,25(8):1021[6]FilipeC,MiguelM,CarlosC,etal.JournalofFoodCompositionandAnalysis,2011.24(3):351
[7]WANGShi-cheng,WANGYan-hong,LIUYan-hui,etal(王世成,王顏紅,劉艷輝,等).Foodscience(食品科學),2013,34(04):183.
[8]SounderajanS,KumarGK,UdasAC.JournalofHazardousMaterials,2010,175(1-3):666.
[9]ZvonimirS,JaroslavaSvarc-Gajic,NikolaM,etal.FoodChemistry,2005,92(4):771
[10]CHANGYIn-fu(常銀甫).PhysicalTestingandChemicalAnalysis(Partb:ChemicalAnalysi)(理化檢驗化學分冊),2010,46(8):970
[11]ZHANGHao,MOHai-zhen,ZHOUQuan-xia,etal(張浩,莫海珍,周全霞,等).Foodscience(食品科學),2010,31(14):211
[12]WANGBing-tao,XIELi-qi,LINYan-kui,etal(王丙濤,謝麗琪,林燕奎,等),ChineseJournalofChromatography(色譜),2011,29(3):223.
[13]HULiang,DO)NGZe-qin,HUANGXIao-han,etal(胡良,董澤琴,黃笑寒,等).ChineseJournalofAnalyticalChemistry(分析化學),2011,39(4):466
[14]2010.ThePeople’sRepublicofChinaNationalStandard(中華人民共和國國家標準)NationalFoodSafetyStsndardDeterminationofseleniuminFoods(食品安全國家標準食品中硒的測定)。


掃描二維碼
版權所有©廣州格丹納儀器有限公司 ![]() 粵公網安備 44011202000628號 粵ICP備14047970號
粵公網安備 44011202000628號 粵ICP備14047970號
分享到:







